
近年来,随着信息技术和生物,化学的快速发展,使用计算机来辅助药物开发的计算机辅助药物设计(Computer-Aided Drug Design, CADD)流程开始被业界广泛接受并投入使用。在整个CADD流程中,虚拟筛选是非常重要的一个内容。
针对一个已知的致病蛋白,需要从十亿级规模的小分子库中,提取出每一个小分子与致病蛋白进行对接构象的计算预测,得到在计算层面两者结合的最佳构象,并对其进行结合能的计算,按照结合能对每一个小分子进行打分,最后按照小分子得分高低来筛选出与该蛋白亲和性高的小分子,再进行下一步的临床试验,这个流程就叫做虚拟筛选。
虚拟筛选在很大程度上加速了整个药物筛选的流程,但是随着小分子数量的增多,小分子数据库的增大,先进行化合物和蛋白质的对接构象采样,再对构象进行打分这一流程显得略有冗杂,而且构象采样也会消耗大量的算力和时间。因此,通过学习局部已有小分子和蛋白质的对接亲和性,来预测分子库中其他小分子的亲和性,这一思路运营而生,业界中也慢慢涌现了许多根据未对接的小分子和蛋白质来预测两者亲和性的模型。
GraphDTA(drug-target affinity)就是一个业界已有且表现较好的亲和性预测模型。
向GraphDTA模型中输入小分子的SMILES式和蛋白质的氨基酸序列,模型使用GNN网络来提取小分子的特征向量,并且使用CNN来提取蛋白质的特征向量,最后根据两者特征向量来计算预测两者间的亲和性。GraphDTA模型结构如下图所示:
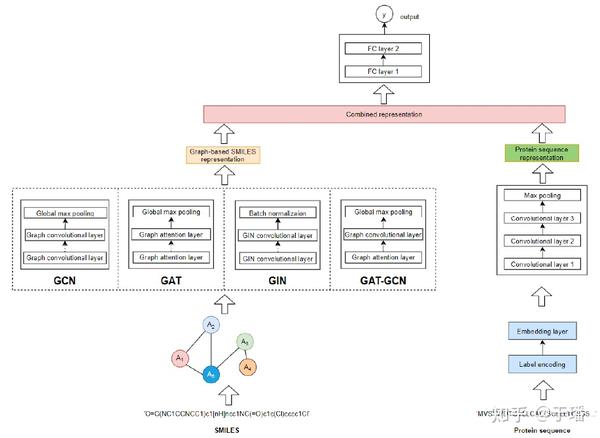
GraphDTA模型结构
模型训练集选取了和DeepDTA相同的数据集,davis和kiba数据集。其中davis数据集中共有442个蛋白质序列和68个小分子的SMILES式,以及它们之间两两结合的Kd/Ki,即亲和性label,共有30056个数据。而kiba数据集中共有229个蛋白质序列和2111个小分子SMILES式,label为KIBA score,共有118254个数据。
GraphDTA中分成两部分来处理输入,对于小分子SMILES式,首先使用openbabel来将SMILES式转换为小分子的3D结构,并且使用基于pytorch的GNN网络模型来处理3D结构,并且生成小分子的特征向量。在模型中提供了4种GNN网络模型,GCN,GAT,GIN和GAT-GCN,分别使用了Graph convolutional layer,Graph attention layer和GIN convolutional layer来形成GNN结构。而在蛋白质部分,则是首先针对输入的蛋白质序列进行embedding,之后将embedding vector输入到三层CNN层的卷积神经网络中去,最后经过一层池化层,得到蛋白质的特征向量。最后将两个特征向量连接起来后通过两层全连接层,计算得到两者的亲和性的预测值。
GraphDTA模型结果如下表所示,在davis数据集中,GraphDTA所提供的四种模型结构的MSE均小于其他模型,而GAT和GIN的CI表现好于其他模型的基准值0.886。在kiba数据集中,GIN,GCN和GAT-GCN的MSE均小于其他模型的最小值,且CI值均高于其他模型。
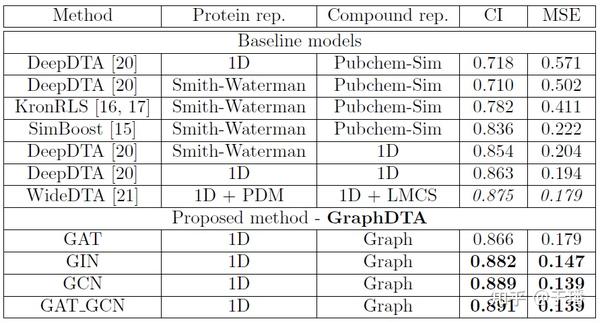
GraphDTA实验结果
值得一提的是,GraphDTA虽然具有很好的表现,但是它仍有改进的空间,在后续的MGraphDTA中,该模型基于GraphDTA模型进行了改造,在处理蛋白质时使用了3种不同的卷积神经网络,在一些数据集上表现也优于GraphDTA。另外,GraphDTA只吸收了蛋白质的一级结构,并没有去学习其3D结构,这显然会导致模型存在信息的缺失,随着alphafold2的发布,能够学习蛋白质的三级结构的模型也渐渐成熟,能否基于这些模型来改进GraphDTA或者创造新的模型结构,将会是接下来蛋白质-小分子亲和性预测的研究主流内容,关于这一话题,有机会的话我们下一次再详细讲解。
作者:于璠
文章来源:知乎
推荐阅读
- LITv2来袭 | 使用HiLo Attention实现高精度、快速度的变形金刚,下游任务均实时
- MindSpore AI科学计算系列(17):AI与“科学”的分工与协作
- 基于卷积神经网络的雾天智能停车位检测:一种新方法
更多嵌入式AI相关技术干货请关注嵌入式AI专栏。欢迎添加极术小姐姐微信(id:aijishu20)加入技术交流群,请备注研究方向。

